Research
Agentic AI Systems for Data Processing & Clinical Risk Prediction
Real-world EHR data are messy: missingness, human error, outliers, and workflow effects can dominate signal and undermine clinical prediction. We develop agentic AI systems that automatically diagnose data quality issues, execute reproducible preprocessing and feature engineering, and generate clinically grounded representations for modeling—while recording provenance and decision rationales for auditability and interpretability. These pipelines include agents that select modeling strategies, configure hyperparameter tuning and validation, and adapt workflows to the dataset and clinical endpoint under explicit constraints (e.g., leakage control and robustness checks). We apply these methods across structured data, unstructured clinical text, and imaging for perioperative risk prediction (including elective spine surgery outcomes) and for predicting problematic opioid use / opioid use disorder.
Clinical NLP for Substance Use Phenotyping
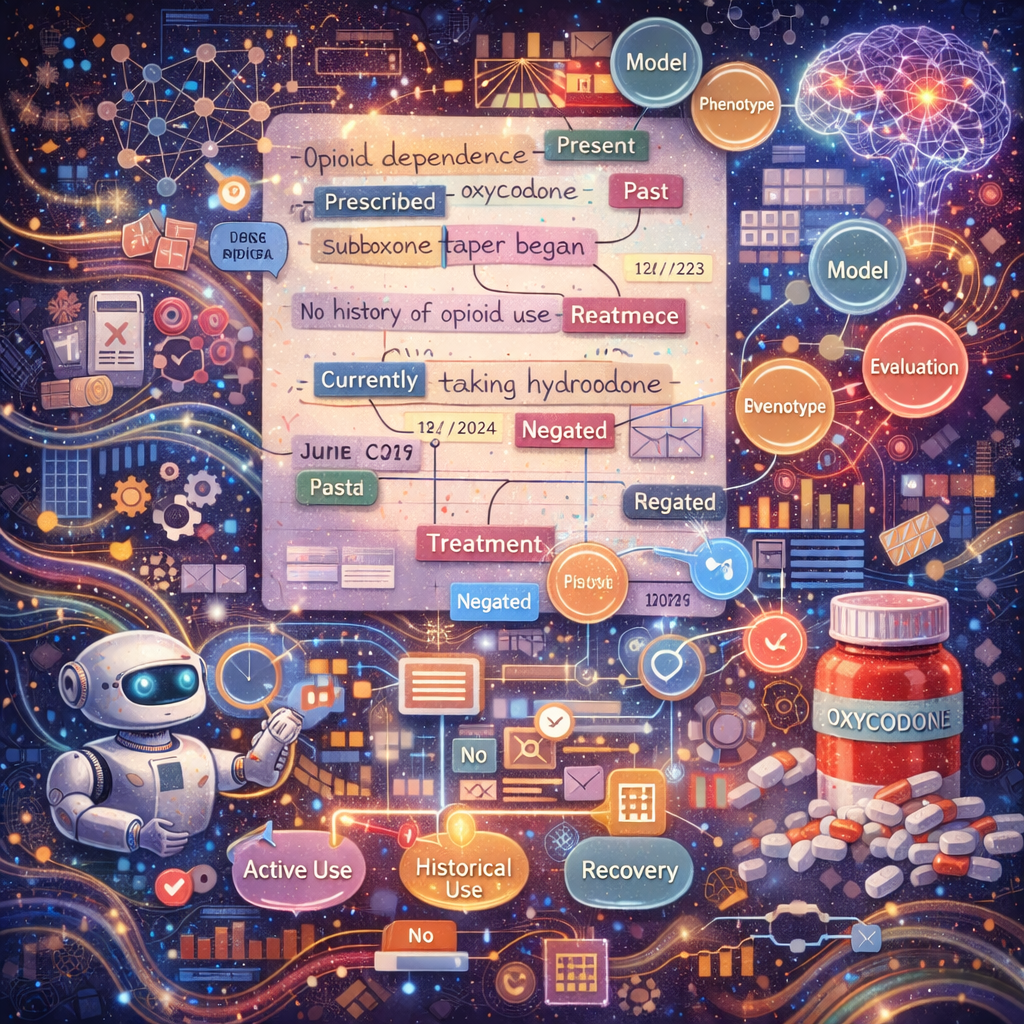
Substance use disorders are often under-coded in structured EHR fields, with key evidence embedded in clinical narratives. We develop clinical NLP and phenotyping frameworks that extract and organize substance-use–related signals from notes and discharge summaries while preserving clinical context (e.g., negation, temporality, uncertainty, and attribution). A major focus is problematic opioid use/opioid use disorder, where we build context-aware annotation and modeling pipelines to distinguish clinically meaningful patterns such as active use vs. historical use, treatment and recovery markers, and severity-related indicators. These phenotypes are designed to be reproducible and evaluation-ready, supporting downstream prediction, cohort discovery, and longitudinal risk profiling.
Knowledge Graphs & Human-AI Collaboration

We build structured frameworks that encode relationships among clinical concepts, data operations, and validation evidence. These knowledge graphs support transparent iteration and discovery of insights, enabling clinicians and data scientists to compare pipeline choices and improve model interpretability through graph-informed, retrieval-augmented interfaces. This work aims to make the AI decision-making process more accessible and auditable for clinical stakeholders.
Modeling Clinical Heterogeneity

Clinical outcomes vary substantially across patient subgroups, yet most models assume homogeneous populations. Our research addresses this by identifying clinically meaningful risk phenotypes, quantifying performance stability across strata, and generating calibrated risk estimates for reliable stratification. This work is particularly relevant for surgical outcomes and substance use disorders, where patient heterogeneity can dramatically affect treatment decisions and risk assessment.
AutoML for Genotype–Phenotype Association Analysis
Complex traits often depend on epistatic (gene-gene) interactions and non-additive genetic effects that standard GWAS approaches miss. We leverage genetic programming and evolutionary algorithms to tackle these challenges. The StarBASE-GP tool we've developed uses genetic programming to evolve interpretable models that evaluate multiple inheritance patterns and detect epistatic interactions, moving beyond additive-only assumptions to improve detection of complex genetic architectures. This work spans addiction genetics, metabolic traits, and model organism studies, with a focus on making interaction findings biologically interpretable and clinically actionable.